Deletions and duplications in the human genome are a major cause for neurodevelopmental disorders such as autism, epilepsy, and intellectual disability. However, not all individuals carrying these deletions and duplications share similar clinical features with high variability in the extent and severity of presenting signs and symptoms. This suggests that these individuals may have other genetic variations ("second hits") that modify the manifestation and the trajectory of the progression of the disorder. In 2010, we analyzed 2,312 individuals with genomic disorders such as 16p11.2 deletion, 16p13.11 deletion, 15q13.3 deletion, 1q21.1 deletion, and 16p12.2 deletion, and found that individuals carrying additional deletions and duplications elsewhere in the genome showed more clinical features than those with only deletion. More recently, in 2018, we analyzed 990 individuals with either rare deletions or single-base pair changes in genes predicted to cause autism, and found that individuals with a higher number of second hits elsewhere in the genome (both single-base pair changes and larger deletions) had more severe clinical features. For example, we found correlations between the number of second hits and IQ and head size for individuals with the 16p11.2 deletion, and found that children with the 16p12.2 deletion who had a strong family history of psychiatric disorders were more likely to have early-onset developmental disorders and a higher number of second hits compared to their parents who carried the same deletion.
The Girirajan lab is currently seeking to understand the biological basis of how the second hits influence the spectrum of clinical symptoms associated with neurodevelopmental disorders, from intellecual disability and autism severity to head circumference and BMI. Our project aims to search for second hits or genetic modifiers within genomes of affected patients and their family members using high throughput, state-of-the-art genomic sequencing techniques. This work will provide a better understanding of the biological mechanisms behind the manifestation of clinical symptoms associated with the genomic disorders. Using sophisticated computational methods such as machine learning, we will be able to map the combination of genetic variants that confer high risk for disease. With the knowledge gained from this project, we hope to lay a foundation to provide a better quality of life to those who suffer from neurodevelopmental disorders.
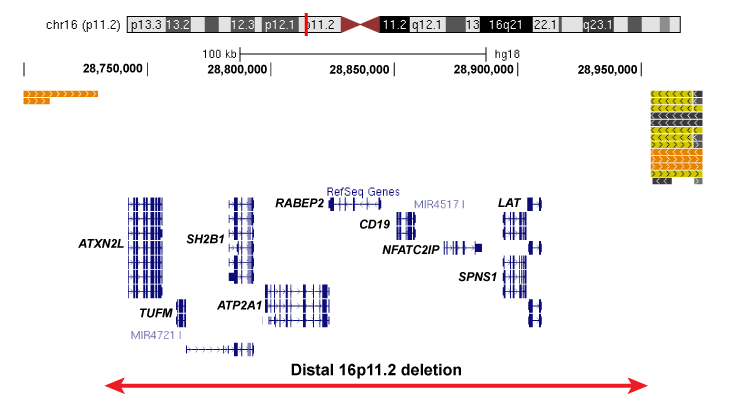
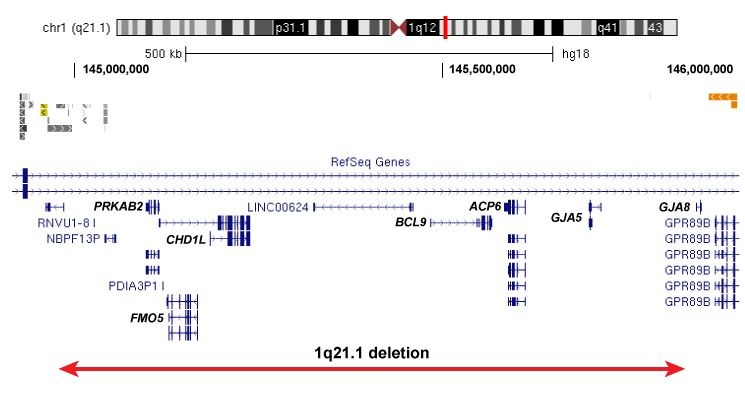
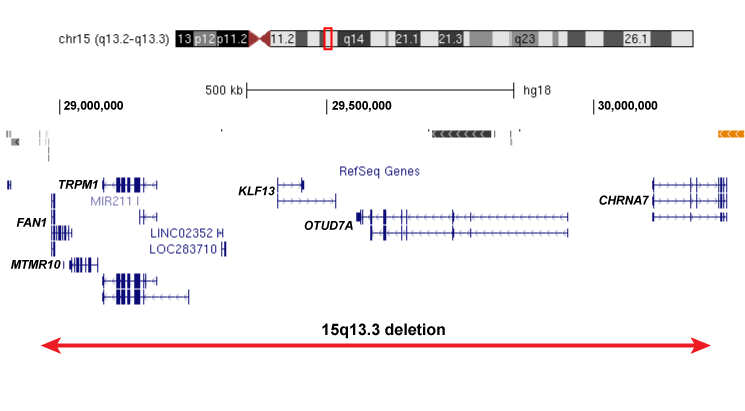
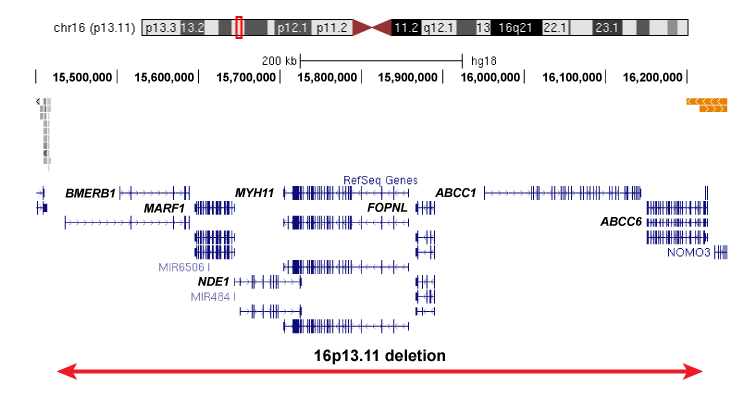
Our work would not be possible without the recruitment of patients for this study. We are currently recruiting individuals with specific rare deletions and their family members for our study, and invite you to participate if your child or loved one of yours has been affected by the following deletions: 1q21.1 deletion,15q13.3 deletion, 16p13.11 deletion, 16p12.1 deletion, distal 16p11.2 deletion.
Please click on the images below to learn more about each of the deletions and to find out more about how to participate in our project:
The Girirajan lab is currently seeking to understand the biological basis of how the second hits influence the spectrum of clinical symptoms associated with neurodevelopmental disorders, from intellecual disability and autism severity to head circumference and BMI. Our project aims to search for second hits or genetic modifiers within genomes of affected patients and their family members using high throughput, state-of-the-art genomic sequencing techniques. This work will provide a better understanding of the biological mechanisms behind the manifestation of clinical symptoms associated with the genomic disorders. Using sophisticated computational methods such as machine learning, we will be able to map the combination of genetic variants that confer high risk for disease. With the knowledge gained from this project, we hope to lay a foundation to provide a better quality of life to those who suffer from neurodevelopmental disorders.
Our work would not be possible without the recruitment of patients for this study. We are currently recruiting individuals with specific rare deletions and their family members for our study, and invite you to participate if your child or loved one of yours has been affected by the following deletions: 1q21.1 deletion,15q13.3 deletion, 16p13.11 deletion, 16p12.1 deletion, distal 16p11.2 deletion.
Please click on the images below to learn more about each of the deletions and to find out more about how to participate in our project:
1q21.1 deletion
15q13.3 deletion
16p13.11 deletion
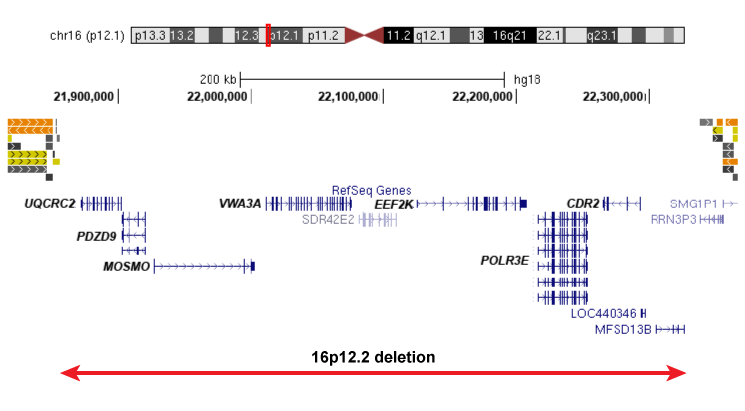
16p12.2 deletion
Distal 16p11.2 deletion