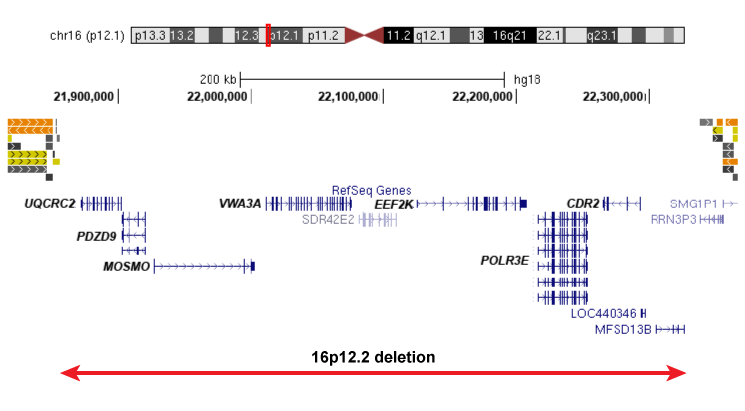
The 16p12.2 deletion (previously termed 16p12.1 deletion) is significantly associated with neurodevelopmental disease, and occurs at a frequency of 1 in 2,000 live births. The primary 16p12.2 deletion spans 7 genes of interest: PDZD9, UQCRC2, C16orf52, VWA3A, EEF2K, POLR3E, and CDR2 (right figure).
Individuals with the deletion have a sensitized genomic background rendering them vulnerable to multiple neurodevelopmental disorders such as intellectual disability, autism, and epilepsy. We are currently studying the 16p12.2 deletion as a paradigm to understand the complex genetics of developmental disorders. We hypothesize that genetic variants near or within the CNV interval and/or at a second site elsewhere in the genome contribute to the phenotypic outcome of disorders association with the deletion. Because the 16p12.2 deletion is inherited from parents in >90% of cases, assessing family history is essential.
A wide array of clinical features has been associated with the 16p12.2 deletion. Affected individuals may exhibit intellectual disability/developmental delay, speech delay, autism-like features, sleep
disorders, hypotonia, craniofacial abnormalities, growth retardation, microcephaly, and cardiac disease. Furthermore, seizure disorders are present and may manifest in several forms including West Syndrome, febrile seizures, or seizure-like episodes. Psychiatric and behavioral abnormalities have also been documented. Individuals carrying the 16p12.2 deletion do not present a common facial gestalt and do not share identical clinical presentation, which indicates that this deletion is non-syndromic.
Most genomic disorders are the result of non-allelic homologous recombination (NAHR) between large (larger than 10kb) and highly similar sequences, called segmental duplications. Several chromosomes are enriched in these types of DNA sequences, such as chromosome 16, most particularly its short arm (p=petite, 16p). This leads to a higher frequency of these rearrangements and therefore genomic disorders associated with this variation.
Study of the 16p12.2 deletion continues to be promising in understanding the variable clinical features associated with the disorder, and will lay a foundation
to provide better treatment methods to patients in the years to come.
We are currently recruiting individuals with the 16p12.2 deletion and their family members for this study, and invite you to participate if your child or loved one has been affected by the deletion. The following link leads to an initial questionnaire, which we will use to obtain some preliminary information about you and your family members who carry the 16p12.2 deletion.
We estimate that the survey will take approximately 15 minutes to complete. We note that participation in this questionnaire is completely voluntary. All answers will be kept strictly confidential, and protected to the maximum extent as required by law.
After you complete the questionnaire, we will contact you directly with further information on our study. Your participation in this study will be very important in understanding the connections between genetic variants and the symptoms identified in individuals carrying the 16p12.2 deletion.
Link to 16p12.2 deletion questionnaire


